
Фенолы
Электрофильное замещение в ароматическом кольце фенолов
Реакции ароматического кольца фенолов протекают по механизму электрофильного ароматического замещения. Гидроксильная группа ориентирует вступление электрофильного реагента в орто- и пара-положения бензольного кольца. Из-за донорного влияния гидроксигруппы (+М эффект) реакции электрофильного замещения в фенолах протекают значительно легче, чем в бензоле и его гомологах. Фенолы и фенолят-анионы вступают во взаимодействие даже с очень слабыми электрофилами, с которыми большинство других ароматических соединений не реагирует – катионами арендиазония ArN2+, нитрозония NO+, иминия R2C=N+H2 и диоксидом углерода (одним из самых слабых электрофилов).
В отличие от бензола галогенирование фенола не требует катализа и протекает при действии молекулярного галогена. При бромировании фенола в водном растворе остановить реакцию на стадии образования монобромпроизводного невозможно, образуется сразу 2,4,6-трибромофенол, который при избытке брома превращается в 2,4,4,6-тетрабромоциклогексадиен-2,5-он. Это происходит из-за того, что в воде фенол частично диссоциирован и галогенированию подвергается фенолят-анион, значительно легче вступающий в реакции электрофильного замещения. Получающийся вначале монобромфенол является более сильной кислотой (-I эффект атома брома) по сравнению с фенолом и, следовательно бромируется быстрее, чем фенол. 2,4-Дибромфенол, в свою очередь, реагирует с бромом легче, чем бромфенол. Как следствие – образование в этой реакции исключительно трибромпроизводного даже при недостатке брома.

Для того чтобы осуществить моногалогенирование фенола, необходимо проводить реакцию в условиях, где диссоциация фенола полностью подавлена. Например, п-бромфенол синтезируют при действии на фенол бромом в среде бромоводородной кислоты, сероугрерода или реакцией фенола с диоксандибромидом в хлороформе.

Нитрование фенола протекает при действии разбавленной азотной кислоты и приводит к двум изомерам – орто- и пара-нитрофенолам. Дальнейшее взаимодействие с концентрированной азотной кислотой дает сначала динитрофенолы и затем 2,4,6-тринитрофенол (пикриновую кислоту). Этот процесс сопровождается значительным смолообразованием из-за окислительного действия азотной кислоты.

Поэтому для синтеза пикриновой кислоты преимущественно применяют нитрование 4-гидроксибензол-1,3-дисульфокислоты, основанное на ипсо-замещении сульфогрупп на нитрогруппы.

Сульфирование фенола осуществляют путем нагревания с концентрированной серной кислотой. Температура проведения реакции решающим образом определяет строение образующихся оксибензолсульфокислот. орто-Изомер, скорость образования которого выше, чем пара-изомера, является доминирующим продуктом, если температура проведения реакции не превышает 100 °С. Его называют кинетическим продуктом. Напротив, при более высоких температурах основным продуктом является пара-изомер, скорость образования которого ниже, но он обладает высокой термодинамической устойчивостью Реакция электрофильного ароматического сульфирования обратима и при нагревании орто-оксибензолсульфокислоты, с серной кислотой выше 100 °С получают пара-изомер – продукт термодинамического контроля реакции.

В отличие от алкилирования фенола по гидроксигруппе, которое идет в щелочной среде, введение алкильных заместителей в ароматическое кольцо фенола протекает при действии галогеналканов, спиртов или алкенов в присутствии катализаторов – минеральных кислот или кислот Льюиса (реакция Фриделя-Крафтса).


орто-Аллилфенолы образуются при нагревании до 200-220 °С аллилфениловых эфиров. Это превращение называют перегруппировкой Кляйзена. Реакция протекает внутримолекулярно и аллильная группа присоединяется к бензольному кольцу γ-углеродным атомом.

Если орто-положения в аллилфениловом эфире заняты, по перегруппировка Кляйзена осуществляется в пара-положение. Так, аллил-2,6-диметилфениловый эфир перегруппировывается при нагревании в 4-аллил-2,6-диметилфенол.

Ацилирование фенола по Фриделю-Крафтсу можно осуществить только в присутствии кислот Льюиса. Фенол взаимодействует с ацетилхлоридом в присутствии хлорида алюминия, давая смесь о- и п-ацетилфенолов (о- и п-гидроксиацетофенонов).

Другой способ получения С-ацилпроизводных фенолов основан на упомянутой выше перегруппировке Фриса. Ариловые эфиры карбоновых кислот при нагревании в присутствии AlCl3 или AlBr3 превращаются в орто- и пара-ацилфенолы. При этом, варьируя температуру и растворитель, можно изменить соотношение изомеров в пользу одного из них. Механизм реакции включает две стадии: 1) межмолекулярное ацилирование бензольного кольца одной молекулы сложного эфира комплексом второй молекулы с кислотой Льюиса; 2) межмолекулярный перенос О-ацильной группы к молекуле фенола.


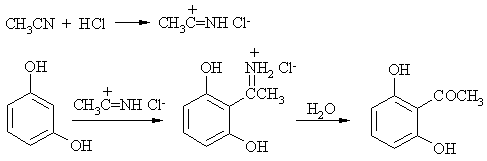
Ацилирование многоатомных фенолов нитрилами в присутствии газообразного хлороводорода носит название реакции Геша. Например, резорцин при действии ацетонитрила в условиях реакции Геша дает 2,6-дигидроксиацетофенон.
Ацилирование фенолов фталевым ангидридом в присутствии серной кислоты или хлорида цинка приводит к образованию фталеинов, многие из которых являются индикаторами (фенолфталеин, тимолфталеин) или красителями (флуоресцеин).

Введение формильной группы в ароматическое кольцо фенолов осуществляется различными методами – для этого применимы реакции Реймера-Тимана, Вильсмайера-Хаака, Гаттермана.
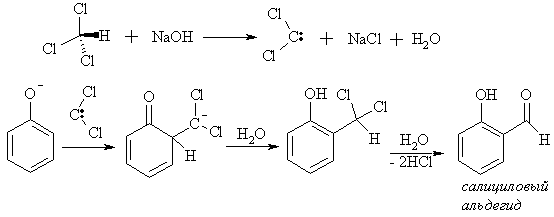
Взаимодействие фенола с хлороформом в щелочной среде (реакция Реймера-Тимана) дает орто-гидроксибензальдегид (салициловый альдегид).

Электрофильной частицей в реакции выступает дихлоркарбен, который генерируется из хлороформа под действием щелочи. Промежуточно образующийся орто-(дихлорметил)фенол гидролизуется в ходе реакции до альдегида. По Реймеру-Тиману формилируется большинство С-замещенных фенолов, нафтолы и многоатомные фенолы, при этом формильная группа вступает всегда в орто-положение относительно гидроксила.
Для формилирования фенолов по Вильсмайеру-Хааку используют замещенные амиды муравьиной кислоты – N,N-диметилформамид и N-метилформанилид, которые в ходе реакции превращают в электрофильные иминиевые соли взаимодействием с хлорокисью фосфора, тионилхлоридом или фосгеном. Последние, хотя и являются слабыми электрофилами, атакуют ароматическое ядро, что приводит к соответствующим иммониевым солям, которые далее гидролизуются до альдегидов. Формильная группа вводится исключительно в пара-положение относительно гидроксильной группы.

Классическое формилирование по Гаттерману-Коху (СО и HCl), протекающее для бензола, нафталина и их гомологов, для фенолов неосуществимо. Однако сам Л. Гаттерман предложил модификацию метода, приемлемую для фенолов, в том числе и многоатомных, и нафтолов. При действии на фенол б/в HCN и газообразного хлороводорода (реакция Гаттермана) образуется пара-гидроксибензальдегид. В некоторых случаях вместо токсичного цианистого водорода используют цианид цинка. Природа электрофильной реагирующей частицы в этой реакции точно не определена.
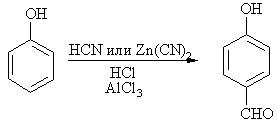
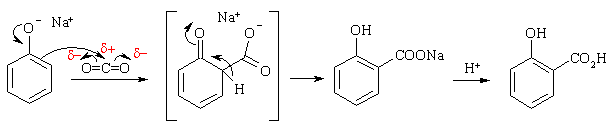
Карбоксилирование (реакция Кольбе)
Нагревание фенолята натрия с диоксидом углерода при давлении в 5 атм приводит к образованию салицилата натрия. Интересно, что при использовании в качестве исходного соединения фенолята калия карбоксильная группа вступает в пара-положение бензольного кольца и образуется пара-гидроксибензоат калия. Электрофилом в реакции Кольбе выступает диоксид углерода, который атакует электроноизбыточные орто- или пара-положения фенолят-аниона. Предполагают, что если в реакционной смеси присутствуют ионы натрия, то за счет комплексообразования происходит стабилизация переходного состояния, ведущего к образованию салициловой кислоты.

Многоатомные фенолы реагируют с диоксидом углерода в более мягких условиях. Например, резорцин карбоксилируется уже при кипячении с раствором карбоната натрия.

Фенолят-ионы взаимодействуют с солями арендиазония в слабощелочной среде с образованием производных азобензола. При этом азосочетание протекает преимущественно по пара-положению фенола, и только если пара-положение занято – диазосотавляющая атакует свободное орто-положение. Фенолы, содержащие сильные электроноакцепторы (например, нитрофенолы) не вступают в эту реакцию. (См. главу Диазосоединения)

Нитрозирование
Катион нитрозония, генерируемый из нитрита натрия и соляной или уксусной кислот селективно атакует пара-положение фенола. Получающийся пара-нитрозофенол существует виде таутомерной смеси с монооксимом п-бензохинона, причем содержание последнего превалирует.

Большое промышленное значение имеет конденсация фенола с формальдегидом, в результате которой получают фенолоформальдегидные смолы (бакелит), которое было разработано Бакелундом. Реакция катализируется кислотами или основаниями и по механизму напоминает одновременно альдольно-кротоновую конденсацию альдегидов и электрофильное замещение в аренах. Так, образующийся из формальдегида при действии кислоты карбокатион атакует орто- или пара-положения молекулы фенола. Получающийся гидроксиметилфенол дегидратируется, давая гидроксифенилметильный карбокатион, который далее реагирует с молекулой фенола или гидроксиметилфенола. Дальнейшая поликонденсация приводит к образованию разветвленного полимера бакелита.

|
|
||
|
|
|
|