
Гетероциклические соединения
Пиридин
Превращения по атому азота
Пиридин – основание средней силы с величиной рКа= 5,2, измеренной в воде (основность алифатических аминов 9-11). Нужно отметить, что в газовой фазе пиридин как основание на 4 порядка сильнее аминов. Пиридин образует кристаллические соли с большинством протонных кислот и часто применяется как основный катализатор или растворитель, способствующий связыванию выделяющихся в ходе той или иной химической реакции кислот.
Как нуклеофил, пиридин реагирует с алкилгалогенидами и другими алкилирующими реагентами по механизму SN2 или SN1 в зависимости от природы субстрата.

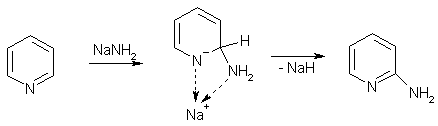
Соли N-алкилпиридиния являются ароматическими соединениями, т.к. неподеленная электронная пара, использующаяся для образования новой связи, не участвует в ароматическом сопряжении. Эти устойчивые вещества проявляют склонность к реакциям с нуклеофилами из-за высокой π-дефицитности, например, аминирование действием амида натрия (реакция Чичибабина) направляется в α-положение

Нуклеофильное гидроксилирование дает α-пиридоны.

При взаимодействии пиридинов с ацилгалогенидами образуются N-ацилиевые соли. Эти соединения мало устойчивы и легко гидролизуются обратно.

Именно по причине низкой устойчивости ацилпиридиниевые соли имеют важное значение для синтетической химии как мягкие ацилирующие реагенты. Например, отметим О-ацилирование кетоенолов – реакция, протекающая по необычному механизму с промежуточным образованием аддукта по α-положению гетероцикла.

При обработке пиридина и его производных надкислотами образуются N-оксиды пиридинов, о свойствах которых будет сказано отдельно.

Реакции по атомам углерода
Взаимодействие с электрофилами
Как было показано выше, пиридин представляет собой π-дефицитное соединение, поэтому взаимодействие с электрофилами для него не характерно, тем более что электрофильные реакции протекают в кислой среде, где во взаимодйствие с электрофилом вступае пиридиниевый ион, обладающий еще большей π-дефицитностью. Эти реакции протекают значительно труднее, чем в бензоле. Пиридин атакуется только сильнейшими электрофилами, причем в весьма жестких условиях. Электрофильное замещение при этом ориентируются в положение 3, что напоминает ориентацию SE2-реакций в нитробензоле (см. главу Нитросоединения). Такая ориентация легко объясняется сравнительной устойчивостью граничных структур, описывающих катионные σ-комплексы, возникающие в результате присоединения электрофила к γ-, β-, и α-положениям пиридинового кольца. Очевидно, что только σ-комплекс II не имеет вклада структуры с положительно заряженным атомом азота.

Как уже было сказано, сначала происходит атака электрофила (или протона) по азоту, что дополнительно пассивирует субстрат, переводя его в катион. Если предотвратить комплексообразование по гетероатому, то реакция атомов ядра с электрофилами протекает более легко.
Нитрование
Нитрование собственно пиридина, особенно нитрующей смесью, когда гетероцикл нацело протонирован, протекает чрезвычайно трудно и практического значения не имеет.

2,6-Диметил- и 2,4,6-триметилпиридины, аминопиридины и пиридоны намного активнее нитруются в форме катионов.

2,6-Дигалогенпроизводные пиридина реагируют легче, т. к. пиридиновый атом азота протонирован в меньшей степени и концентрация свободного основания больше. Нитрование протекает легче в случае использования апротонного нитрующего реагента, например тетрафторбората нитрония:

Сульфирование
Сульфирование пиридина серной кислотой протекает несколько легче, чем нитрование, но тоже возможно лишь в жестких условиях. При еще более высоких температурах возможна перегруппировка в 4-сульфокислоту.

Любопытный продукт может быть получен при сульфировании 2,6-ди-трет-бутилпиридина. Само сульфирование протекает особенно легко, т. к. объемные заместители препятствуют комплексообразованию SO3 по атому азота; при нагревании образуется циклический сульфон.

Пиридины могут быть прогалогенированы. Ввести иод в кольцо пиридина с удовлетворительным выходом не удается, однако бром- и хлорпиридины синтезируются весьма просто.

N-Оксиды пиридина проявляют большую активность в реакциях с электрофилами, чем сам пиридин. Существуют два типа электрофильного замещения в N-оксидах: первый вариант (без добавления нуклеофила) приводит, в основном, к N-оксидам 4-замещенных пиридинов. Это на первый взгляд странное обстоятельство связано с замечательной электронной природой N-оксидной функции, которая может быть одновременно не только акцептором, но и донором электронов. Это может быть видно на следующей схеме:

Поэтому N-оксид любого замещенного пиридина, имеющего свободным положение 4, можно пронитровать дымящей HNO3 с хорошим выходом.

Другой подход к использованию N-оксидов – проведение реакции в присутствии слабых нуклеофилов, которые могут входить в состав реагента, например, ацетилнитрата. При этом N-оксид превращается в неароматический аддукт, который и атакуется электрофилом. Можно сказать, что пиридиновый атом азота на время становится донором электронов. Реакция позволяет получить хорошие выходы 3-нитро- и 3,5-динитропиридинов.

Аналогично происходит вступление в положение 5 еще одной нитрогруппы

Более характерными превращениями пиридина являются реакции нуклеофильного замещения. Замещение атома водорода на аминогруппу протекает под действием амида натрия при нагревании и всегда ориентируется в положение 2. Превращение имеет сложный механизм: в ходе реакции выделяются водород, гидрид натрия, аммиак и другие продукты. Тот факт, что водород в положении 4 не замещается, объясняют предварительной координацией атома натрия с пиридиновым азотом.
Обработка пиридинов свежеплавленным едким кали приводит к гидроксилированию до α-пиридонов (α- и γ-гидроксипиридоны существуют в более устойчивой оксо-форме).

Реакции нуклеофильного замещения галогена в пиридине протекают по таким же двум альтернативным механизмам, как и в галогенаренах (см. главу Нуклеофильное замещение в ароматическом кольце) – присоединение-отщепление (АЕ) и отщепление-присоединение (ЕА). Реакция АЕ (присоединение нуклеофила с образованием анионного σ-комплекса и отщепление уходящей группы) протекают преимущественно по атомам 2, 4 и 6, в которых локадизован больший положительный заряд. Кроме того, атом азота участвует в делокализации отрицательного заряда соответствующих σ-комплексов, как нитрогруппа в случае нитрохлорбензолов. Наиболее стабильными являются анионные σ-комплексы I и III.

С помощью реакции типа нуклеофильного АЕ в молекулу пиридина можно ввести самые разнообразные заместители. Вот несколько примеров:

N-оксиды пиридина и N-алкилпиридиниевые соли вступают в те же реакции легче самого пиридина. Особенно интересны N-оксиды α-галогенпиридинов, которые при действии некоторых нуклеофилов сразу превращаются в конденсированные биядерные гетероциклы, в образовании которых участвует N-оксидная группа.

Если галоген находится в положении 3, то реакция пиридинов с нуклеофилами чаще идет по механизму ЕА через промежуточное образование гетероаналога дегидробензола – так называемого гетарина.

Свободнорадикальные реакции
При действии атомарных хлора и (при более высоких температурах) брома на пиридин происходит свободнорадикальное галогенирование, которое, в отличие от электрофильного, ориентируется в положения 2 и 6.

Для препаративных целей имеют значение реакции пиридина с нуклеофильными радикалами (реакция Миниши). Источниками радикалов служат различные органические соединения в присутствии перекисей и солей железа(II), катион которого служит в качестве переносчика электронов.

Механизм реакции включает стадии гомолитического разложения перекиси, превращения реагента в свободный радикал, его присоединение к пиридину и последующую ароматизацию.

Таким путем в положение 2 пиридина можно ввести гидроксиметильную группу, диалкиламидную и многие другие функциональные группы.
|
|
||
|
|
|
|